Diversity analysis
Packages
library(phyloseq)
library(ggpubr)
library(tidyverse)
theme_set(theme_bw())
Load phyloseq object
For detail on how to get a phyloseq object please see chapter ‘From Qiime2 into R’.
# reading in a previously saved phyloseq object
physeqB.flt <- readRDS('physeqB.flt')
physeqB.flt
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 4932 taxa and 136 samples ]
## sample_data() Sample Data: [ 136 samples by 38 sample variables ]
## tax_table() Taxonomy Table: [ 4932 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 4932 tips and 4931 internal nodes ]
Alpha diversity
Pre-processing of phyloseq object
#filter
`%notin%` = Negate(`%in%`)
physeqB.flt.norefs <- prune_samples(sample_data(physeqB.flt)$Name %notin% c("41AutoclavedControlT0","42AutoclavedControlT0","43AutoclavedControlT0","44AutoclavedControlT0"), physeqB.flt)
physeqB.flt.norefs <- prune_samples(sample_data(physeqB.flt.norefs)$Treatment != "Reference", physeqB.flt.norefs)
physeqB.flt.norefs <- prune_samples(sample_data(physeqB.flt.norefs)$Treatment != "Blank", physeqB.flt.norefs)
physeqB.flt.norefs <- phyloseq::prune_taxa(taxa_sums(physeqB.flt.norefs) != 0, physeqB.flt.norefs)
# checking minimum sample size
min(colSums(otu_table(physeqB.flt.norefs)))
## [1] 9214
# rarefy
rare_Bac <- phyloseq::rarefy_even_depth(physeqB.flt.norefs, sample.size = min(colSums(otu_table(physeqB.flt.norefs))), rngseed = TRUE, replace = TRUE, trimOTUs = TRUE)
# check rarefied phyloseq object
rare_Bac
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 4929 taxa and 128 samples ]
## sample_data() Sample Data: [ 128 samples by 38 sample variables ]
## tax_table() Taxonomy Table: [ 4929 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 4929 tips and 4928 internal nodes ]
#create diversity indices
ps.div <- phyloseq::estimate_richness(rare_Bac)
# add sample names
ps.div$`#SampleID` <- phyloseq::sample_names(physeqB.flt.norefs)
# add diversity indices to phyloseq object
meta.df <- data.frame(sample_data(physeqB.flt.norefs))
meta.df <- meta.df %>% rownames_to_column("#SampleID") %>%
left_join(ps.div, by = "#SampleID") %>% column_to_rownames("#SampleID")
# add pilou evenness (see calculation in "Numerical Ecology with R - Bocard et al")
meta.df$pielou_ev <- ps.div$Shannon/log(ps.div$Observed)
# replace metadata with temp meta.df which includes the diversity indices
physeqB.flt.norefs@sam_data <- sample_data(meta.df)
Plotting
Colors
#manually created with help of https://htmlcolorcodes.com/
cols <- c("#F9E79F", "#FDEBD0", "#F6DDCC", "#E5E8E8", "#CCD1D1", "#AAB7B8", "#707B7C", "#566573","#82E0AA", "#641E16", "#8E44AD")
cols_noheatH2O2 <- c("#F9E79F", "#E5E8E8", "#CCD1D1", "#AAB7B8", "#707B7C", "#566573","#82E0AA", "#641E16", "#8E44AD")
cols_ref <- c("#F9E79F", "#FDEBD0", "#F6DDCC", "#E5E8E8", "#CCD1D1", "#AAB7B8", "#707B7C", "#566573", "#82E0AA","#641E16", "#8E44AD", "#E74C3C")
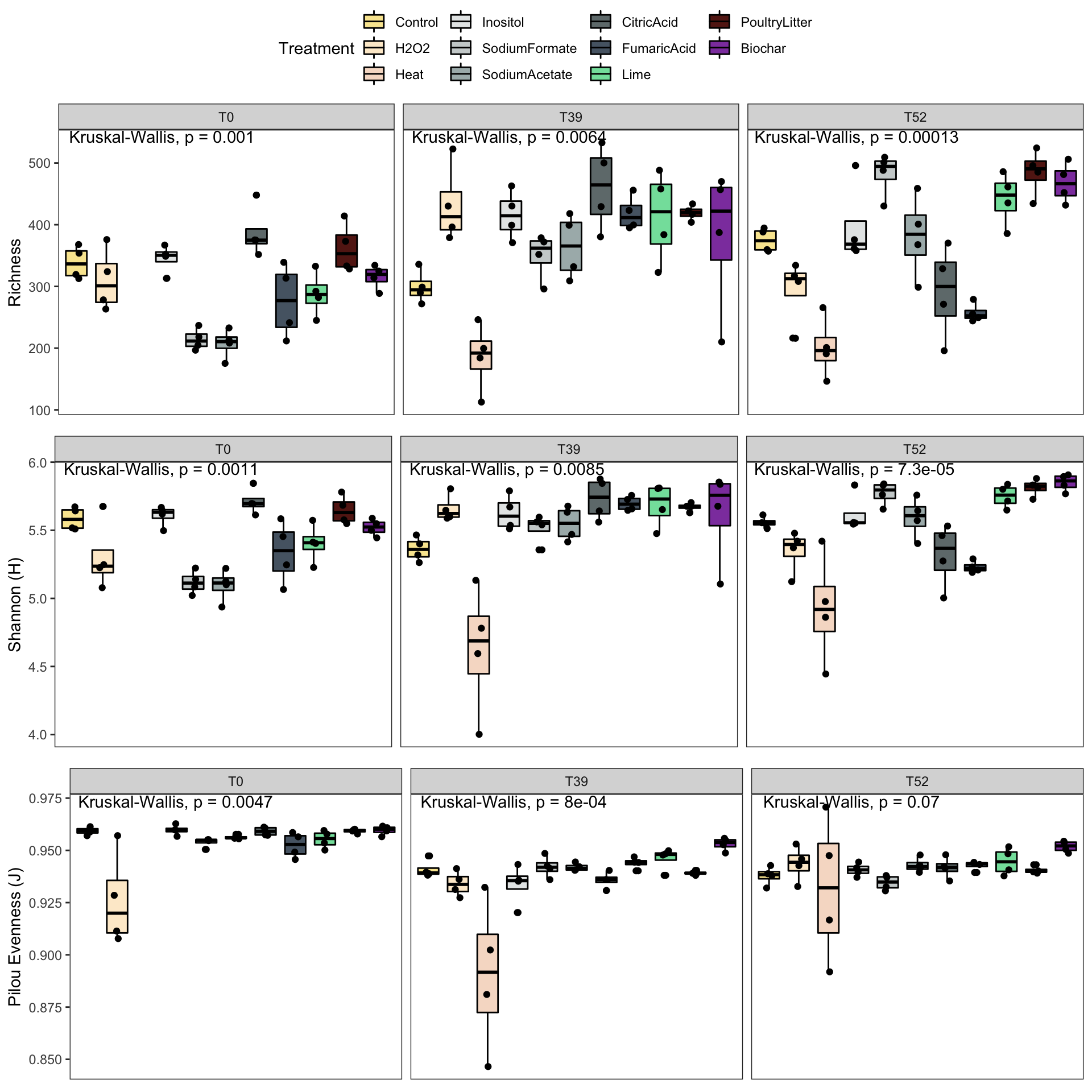
Richness
p1 <- data.frame(phyloseq::sample_data(physeqB.flt.norefs)) %>%
ggpubr::ggboxplot(., x = "Treatment", y = "Observed", facet.by = "Week", fill = "Treatment", ylab = "Richness", xlab = "Treatment", palette = cols) +
theme_bw() +
xlab("") +
theme( panel.grid.major = element_blank(), panel.grid.minor = element_blank() ) +
geom_jitter(width = 0.1) +
# theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
theme(axis.title.x=element_blank(),
axis.text.x=element_blank(),
axis.ticks.x=element_blank()) +
ggpubr::stat_compare_means(method = "kruskal",label.x = 2)
Shannon
# Shannon
p2 <- data.frame(phyloseq::sample_data(physeqB.flt.norefs)) %>%
ggpubr::ggboxplot(., x = "Treatment", y = "Shannon", facet.by = "Week", fill = "Treatment", ylab = "Shannon (H)", xlab = "Treatment", palette = cols) +
theme_bw() +
xlab("") +
theme( panel.grid.major = element_blank(), panel.grid.minor = element_blank() ) +
geom_jitter(width = 0.1) +
# theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
theme(axis.title.x=element_blank(),
axis.text.x=element_blank(),
axis.ticks.x=element_blank())+
ggpubr::stat_compare_means(method = "kruskal",label.x = 2)
Pielou’s evenness
# Pielou
p3 <- data.frame(phyloseq::sample_data(physeqB.flt.norefs)) %>%
ggpubr::ggboxplot(., x = "Treatment", y = "pielou_ev", facet.by = "Week", fill = "Treatment", ylab = "Pilou Evenness (J)", xlab = "Treatment", palette = cols) +
theme_bw() +
xlab("") +
theme( panel.grid.major = element_blank(), panel.grid.minor = element_blank() ) +
geom_jitter(width = 0.1) +
# theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
theme(axis.title.x=element_blank(),
axis.text.x=element_blank(),
axis.ticks.x=element_blank())+
ggpubr::stat_compare_means(method = "kruskal",label.x = 2)
All plots together
pdf(NULL)
g1 <- ggpubr::ggarrange(p1, p2, p3, common.legend = TRUE, ncol = 1)
x = dev.off()
g1
Beta diversity
PCA
# filter and phyloseq object from alphadiversity chunk: "physeqB.flt.norefs"
# create a new phyloseq object to be less error-prone for downstream analyis
physeqPCA <- physeqB.flt.norefs # excluding Heat, H2O2, reference and blank
# normalisation and transform using the microbiome package
# I chose a centred log-ratio transform and a principle component analysis in a euclidean space.
physeqPCA <- microbiome::transform(physeqPCA, "clr")
#ordination
ordination <- phyloseq::ordinate(physeqPCA , "RDA")
p1 <- phyloseq::plot_ordination(physeqPCA, ordination , color = "Treatment", shape = "Week") +
geom_point(aes(size = Dieldrin_trans)) +
ggtitle("PCA (clr transform)") +
# scale_color_manual(values = cols_noheatH2O2)
# OR
scale_color_manual(values = cols_ref)
# p1
# ggsave("PCA.png", height=6, width=7.5, units='in', dpi=600)
knitr::include_graphics("./PCA.png")

RDA
# PERCENT FILTERVALUE FOR ALL RDAs
pcnt <- 0.25
# FORMULA FOR ALL RDAs
form <- formula(~ Treatment+Week)
# labels for arrows
# pos for arrows
xarw <- 5
yarw <- 5
# arrow length map
larw <- 1.5
# arrow length map for chromosol rda
larwch <- 1
# fixed width of x axis
xaxis = c(-6.75, 6.5)
# position of annotation
textposx = c(-4.4)
# arrow color
arrowcolor <- "grey13"
physeqB.flt.norefs <- prune_samples(sample_data(physeqB.flt)$Treatment != "Reference", physeqB.flt)
physeqB.flt.norefs <- prune_samples(sample_data(physeqB.flt.norefs)$Treatment != "Blank", physeqB.flt.norefs)
physeqB.flt.norefs <- prune_samples(sample_data(physeqB.flt.norefs)$Treatment != "Heat", physeqB.flt.norefs)
physeqB.flt.norefs <- prune_samples(sample_data(physeqB.flt.norefs)$Treatment != "H2O2", physeqB.flt.norefs)
physeqB.flt.norefs <- phyloseq::prune_taxa(taxa_sums(physeqB.flt.norefs) != 0, physeqB.flt.norefs)
physeqPCA <- physeqB.flt.norefs # excluding Heat, H2O2, reference and blank
# normalisation and transform
physeqPCA <- microbiome::transform(physeqPCA, "clr")
#ordination
ordination <- phyloseq::ordinate(physeqPCA, "RDA", "bray", formula = form )
p2 <- phyloseq::plot_ordination(physeqPCA, ordination , color = "Treatment", shape = "Week") +
geom_hline(yintercept=0, linetype="dashed", color = "gray") +
geom_vline(xintercept=0, linetype="dashed", color = "gray") +
geom_point(aes(size = Dieldrin_trans)) +
#scale_colour_gradient(low = colourlow, high = colourhigh) +
ggtitle("RDA Abundances (clr) ~ Treatment+Week ") +
scale_color_manual(values = cols_noheatH2O2)
# Now add the environmental variables as arrows to either of these p1 or p2
arrowmat <- vegan::scores(ordination, display = "bp")
arrowmat <- data.frame(arrowmat)
#rownames(arrowmat) <- arrowlabel
# Add labels, make a data.frame
arrowdf <- data.frame(labels = rownames(arrowmat), arrowmat)
# Define the arrow aesthetic mapping
arrow_map <- aes(xend = larw * RDA1,
yend = larw * RDA2,
x = 0,
y = 0,
shape = NULL,
color = NULL,
label = labels)
label_map <- aes(x = xarw * RDA1,
y = yarw * RDA2,
shape = NULL,
color = NULL,
label = labels)
arrowhead = arrow(length = unit(0.02, "npc"))
p2 <- p2 +
geom_segment(
mapping = arrow_map,
size = .5,
data = arrowdf,
color = arrowcolor,
arrow = arrowhead) +
geom_text(
mapping = label_map,
size = 4,
data = arrowdf,
show.legend = FALSE)
# p2
# save figure for publication
# ggsave("RDA.png", height=6, width=7.5, units='in', dpi=600)
knitr::include_graphics("./RDA.png")

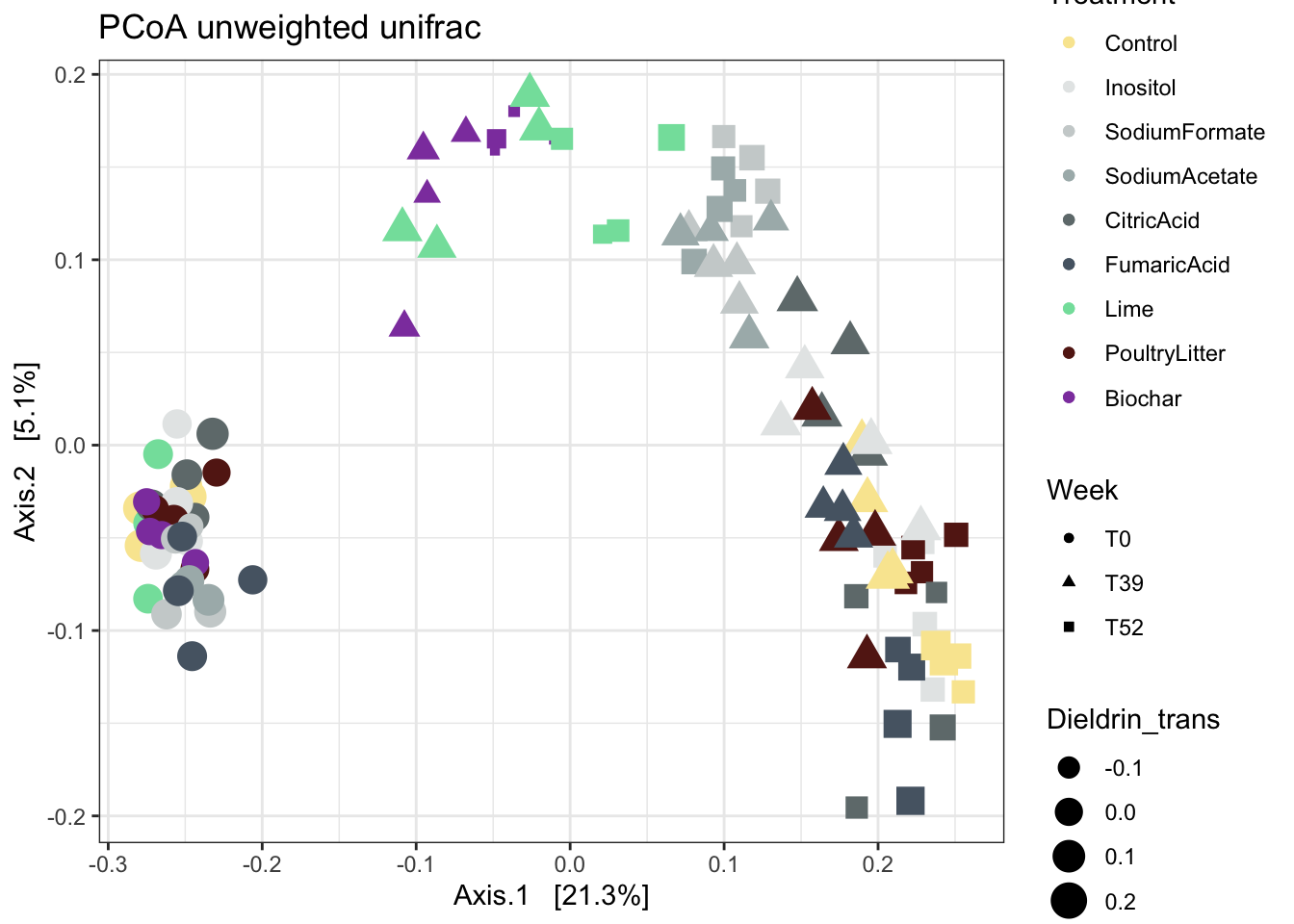
unweighted unifrac
physeqPCA <- physeqB.flt.norefs
#ordination
ordination <- phyloseq::ordinate(physeqPCA , "PCoA", distance = 'unifrac')
p1 <- phyloseq::plot_ordination(physeqPCA, ordination , color = "Treatment", shape = "Week") +
geom_point(aes(size = Dieldrin_trans)) +
ggtitle("PCoA unweighted unifrac") +
# scale_color_manual(values = cols_noheatH2O2)
# OR
scale_color_manual(values = cols_noheatH2O2)
p1
Note: This code is an amalgamation from various sources. Apart from putting it together into a pipeline I do not take credit for it.