Phylofactor differential abundance analysis
Resources
Phylofactor tutorial https://docs.wixstatic.com/ugd/0119a1_099ae20df8424af9a38585dcebc0d45a.pdf
Publication Washburne, A. D., Silverman, J. D., Morton, J. T., Becker, D. J., Crowley, D., Mukherjee, S., … Plowright, R. K. (2019). Phylofactorization: a graph partitioning algorithm to identify phylogenetic scales of ecological data. Ecological Monographs, 89(2), e01353. https://doi.org/10.1002/ecm.1353
Packages
library(phylofactor)
library(phyloseq)
library(ggpubr)
library(tidyverse)
library(ggtree)
Load functions
Phylofactor summary by ASV
Function to summarise all phylofactors into a dataframe including taxonomy details and model coefficients
## phyfacsummary function ##
## Enter the phylofactor file and the phyloseq object to get a dataframe
## with model results, abundances and taxa info
## the third function input is '4' or '5', 4 is used when the
## phylofactor model setting was set to 'F', otherwise 5
## p adjusted method = fdr
phyfacsummary <- function(phylofactorfile, phyloseqobject, Var_or_F) {
datalist = list()
factornumber <- nrow(phylofactorfile$factors)
taxadf <- as.data.frame(tax_table(phyloseqobject)@.Data)
taxadf <- unite(taxadf, taxonomy, sep = ";", remove = TRUE, na.rm = FALSE) %>% rownames_to_column("OTU")
otudf <- data.frame(Abundance = rowSums(as.data.frame(otu_table(phyloseqobject)@.Data))) %>% rownames_to_column("OTU")
for (i in 1:factornumber) {
smry <- phylofactor::pf.summary(phylofactorfile, taxadf, factor=i)
td <- phylofactor::pf.tidy(smry)
factorsgroup <- data.frame(smry$group1) %>% dplyr::select(IDs.otuIDs,IDs.TaxaIDs) %>%
dplyr::rename(OTU = IDs.otuIDs) %>% dplyr::rename(Taxa_ID = IDs.TaxaIDs) %>%
mutate(Phylofactor = i) %>%
mutate('Pr(>F)' = phylofactorfile$factors[[i,Var_or_F]] )
ncoefs <- nrow(data.frame(td$Coefficients))
for(c in 1:ncoefs) { # Head of for-loop
new <- rep(td$Coefficients[[c]], nrow(factorsgroup)) # Create new column
factorsgroup[ , ncol(factorsgroup) + 1] <- new # Append new column
colnames(factorsgroup)[ncol(factorsgroup)] <- paste0(rownames(data.frame(td$Coefficients[c]))) # Rename column name
}
datalist[[i]] <- factorsgroup
}
phylofactorsummary <- dplyr::bind_rows(datalist)
phylofactorsummary$`Pr(>F)adj.` <- p.adjust(phylofactorsummary$`Pr(>F)`, "fdr")
return(phylofactorsummary %>% left_join(otudf, by = "OTU") %>% as_tibble() %>% separate(Taxa_ID, sep=";", c("Kingdom","Phylum","Class","Order","Family","Genus","Species")))
}
Phylofactor summary by factor
# Function to create a dataframe containing all factors including coefficents .
# Makes sure to have the correct phyloseq object which contains the data used to create the phylofactor model
myfactoroverviewfunction <- function(phylofactorobject, phyloseqobject) {
# create a factor dataframe (standard output is without coefficients which will be added with this function)
factors <- data.frame(phylofactorobject$factors)
taxadf <- as.data.frame(tax_table(phyloseqobject)@.Data)
taxadf <- unite(taxadf, taxonomy, sep = ";", remove = TRUE, na.rm = FALSE) %>% rownames_to_column("OTU")
# Create a dataframe with coefficients
ncoefs <- nrow(factors)
df <- data.frame(Factors = rownames(factors))
for(c in 1:ncoefs) { # Head of for-loop
smry <- phylofactor::pf.summary(phylofactorobject, taxadf, factor=c)
td <- phylofactor::pf.tidy(smry)
#df[c,1] <- paste("Factor", 1)
# add coefficient for all factor variables
coefs <- nrow(data.frame(td$Coefficients))
for(v in 1:coefs) { # Head of for-loop
new <- rep(td$Coefficients[[v]], ) # Create new column
df[c , v+1 ] <- new # Append new column
colnames(df)[(v+1)] <- paste0(rownames(data.frame(td$Coefficients[v]))) # Rename column name
}
}
# combine the factor dataframe with coefficient dataframe
factors <- (factors %>% rownames_to_column("Factors") %>% left_join(df))
return(factors)
}
Bin colour function
##### Function for randomly selecting colors from two sets of colours.
## The output of this function is required for the binplotfunction.
# This function allocates a bright and a grey colour set to bins based on bin size.
# within these two colour pools colours are selected at random for phylofactor bins ####
# Seven inputs are required
# Inputs are the phylofactor object (output of a PhyloFactor function) and a phyloseq object (which was used for phylofactor inputs otus and metadata) and the name of the orderfactor which was used for modelling the abundances
# The two colour-sets need to be vectors containing colours which can be any number of colours as long as the total number of colours are identical for both sets
# The first set of colours should be a bright set of colours to highlight larger bins
# the second set of colours is a grey set. These are needed for simplification and so one can identify important bins in the legend.
# The sixed input is a numeric value (0-1) to regulate the size of bins to be coloured. The larger this value results in only larger bins get colours.
# The last input is either 'min' or 'max'. This determines where bin sizes are calculated for deciding on colours. Either at the beginning or at the end of the figure.
# This function requires that the PhyloFactor function was run - using one (not multiple) explanatory variable in the model
mycolorfunction <- function(phylofactorobj, phyloseqobj, orderfactor, brightcols, greycols, brightbinfactor, minmax) { #orderfactor = the explanatory variable used in model
require(rlang)
# extract bins from phylofactor object
bin.projection <- phylofactor::pf.BINprojection(phylofactorobj, rel.abund=T, prediction = T)
bin.observed <- bin.projection$Data
meta.df <- data.frame(sample_data(phyloseqobj)) %>% arrange(Week, SampleNo)
taxadf <- as.data.frame(tax_table(phyloseqobj)@.Data)
taxadf <- unite(taxadf, taxonomy, sep = ";", remove = TRUE, na.rm = FALSE) %>% rownames_to_column("OTU")
# prepare bin table, extract data from phyloseq object and combine
bins <- meta.df %>% dplyr::select(Week, Treatment, {{orderfactor}}) %>%
rownames_to_column("SampleID") %>%
left_join(data.frame(t(bin.observed)) %>%
rownames_to_column("SampleID")) %>%
dplyr::select(-Bin.1) %>%
dplyr::arrange({{orderfactor}}) %>%
pivot_longer(cols = starts_with("Bin"))
# Create a vector with colors, depending on bin size
set.seed(123)
data <- bins %>% dplyr::filter({{orderfactor}} == minmax(bins %>% select({{orderfactor}}))) # filter to the smallest variable value, determines where to filter values
binrows <- nrow(data) # loop number
colvec <- vector("character") # open a vector
for(row in 1:binrows) { # for loop to fill vector
boom <- ifelse( ( data[row,]$value > (brightbinfactor * sum(data$value))), base::sample(brightcols, size = 1), base::sample(greycols, size = 1))
colvec <- c(colvec, boom)
}
names(colvec) <- data$name
return(colvec)
}
Binplot function
##### Function for plotting observed and predicted phylofactor bins ####
# Three inputs are required
# Inputs are a phylofactor object (output of a PhyloFactor function) and a phyloseq object (which was used for phylofactor inputs otus and metadata) and the name of the orderfactor which was used for modelling the abundances
# This function requires that the PhyloFactor function was run - using one (not multiple) explanatory variable in the model
binplotfunction <- function(phylofactorobj, phyloseqobj, orderfactor) {
require(rlang)
# Prepare bin data for plotting
# load meta data from phylo
meta.df <- data.frame(sample_data(phyloseqobj)) %>% arrange(Week, SampleNo)
bin.projection <- phylofactor::pf.BINprojection(phylofactorobj, rel.abund=T)
bin.observed <- bin.projection$Data
# observed values
bins <- meta.df %>% dplyr::select(Week, Treatment, {{orderfactor}}) %>%
rownames_to_column("SampleID") %>%
left_join(data.frame(t(bin.observed)) %>%
rownames_to_column("SampleID")) %>%
dplyr::select(-Bin.1) %>%
dplyr::arrange({{orderfactor}}) %>%
pivot_longer(cols = starts_with("Bin"))
# sorting bins by bin number (i.e. make bin name a factor) so legend appears in correct order
data <- bins %>% dplyr::filter({{orderfactor}} == min(bins %>% select({{orderfactor}})))
bins$name <- factor(bins$name, levels = data$name)
# mutate(plotme = factor({{orderfactor}}) ) # create a leveled factor for plotting in correct order
# only needed if manual ordering is required
p <- ggplot(bins, aes(x={{orderfactor}}, y=value, group=name,fill=name)) +
geom_area(position="fill") +
ggpubr::rotate_x_text(angle = 90) +
ylab("Relative abundanced (Observed)") +
theme(legend.title = element_blank()) +
scale_fill_manual(values = colvec) +
guides(fill = guide_legend(nrow = 5, byrow = TRUE))
# repeat for predicted values
bin.projection <- phylofactor::pf.BINprojection(phylofactorobj, rel.abund=T, prediction = T)
bin.predicted <- bin.projection$Data
bins <- meta.df %>% dplyr::select(Week, Treatment, {{orderfactor}}) %>%
rownames_to_column("SampleID") %>%
left_join(data.frame(t(bin.predicted)) %>%
rownames_to_column("SampleID")) %>%
dplyr::select(-Bin.1) %>%
dplyr::arrange({{orderfactor}}) %>%
pivot_longer(cols = starts_with("Bin"))
# sorting bins by bin number (i.e. make bin name a factor) so legend appears in correct order
data <- bins %>% dplyr::filter({{orderfactor}} == min(bins %>% select({{orderfactor}})))
bins$name <- factor(bins$name, levels = data$name)
# mutate(plotme = factor({{orderfactor}}) ) # create a leveled factor for plotting in correct order
p2 <- ggplot(bins, aes(x={{orderfactor}}, y=value, group=name,fill=name)) +
geom_area(position="fill") +
ggpubr::rotate_x_text(angle = 90) +
ylab("Relative abundanced (Predicted)") +
theme(legend.title = element_blank()) +
scale_fill_manual(values = colvec) +
guides(fill = guide_legend(nrow = 5, byrow = TRUE))
p3 <- ggarrange(p, p2, common.legend = TRUE)
return(p3)
}
Tree colour function
Function to create a vector of randomly selected colors from two sets of pre-defined colours. This vector can be used to colour taxonomic levels in a phylogenetic tree
##### Function for randomly selecting colors from two sets of colours.
# it randomly selects colors from two sets of colours.
# a bright and grey set of colours is selected based on bin size.
# within the colour pools colours are selected at random for each phylofactor bin ##
##### Function for randomly selecting colors from two sets of colours.
# it randomly selects colors from two sets of colours.
# a bright and grey set of colours is selected based on bin size.
# within the colour pools colours are selected at random for each phylofactor bin ##
mytreecolorfunction <- function(phyloseqobj, brightcols, greycols, prevelancethreshold, seed, taxalevel = "Phylum") {
#orderfactor = the explanatory variable used in model
# extract bins from phylofactor object
prevelancedf = apply(X = phyloseq::otu_table(phyloseqobj),
MARGIN = 1,
FUN = function(x){sum(x > 0)})
prevelancedf = data.frame(Prevalence = prevelancedf,
TotalAbundance = taxa_sums(phyloseqobj),
phyloseq::tax_table(phyloseqobj))
prevelancedf <- plyr::ddply(prevelancedf, taxalevel, function(df1){
data.frame(mean_prevalence=mean(df1$Prevalence),total_abundance=sum(df1$TotalAbundance,na.rm = T),stringsAsFactors = F)
})
# Create a vector with colors, depending on prevalence
set.seed(seed)
phyla <- nrow(prevelancedf) # loop number
colvec <- vector("character") # open a vector
for(phylum in 1:phyla) { # for loop to fill vector
boom <- ifelse( ( prevelancedf[phylum,]$total_abundance > prevelancethreshold), base::sample(brightcols, size = 1), base::sample(greycols, size = 1))
colvec <- c(colvec, boom)
}
names(colvec) <- as.vector(prevelancedf %>% dplyr::select(taxalevel))[,1]
zerovec <- c("grey")
names(zerovec) <- "No colour"
colvec <- c(zerovec, colvec)
return(colvec)
}
colour sets
# colour set 1 for mycolorfunctions
brightcols <-c("#E6B0AA","#F5B7B1", "#D7BDE2", "#D2B4DE","#A9CCE3", "#AED6F1", "#A3E4D7","#A2D9CE", "#A9DFBF", "#ABEBC6", "#F9E79F", "#FAD7A0","#F5CBA7", "#EDBB99", "#C0392B", "#E74C3C","#9B59B6", "#2980B9", "#3498DB", "#1ABC9C", "#27AE60","#2ECC71","#F1C40F","#F39C12","#E67E22","#CA6F1E")
# colour set 2 for mycolorfunctions
greycols <-c("#E5E8E8","#CCD1D1", "#B2BABB", "#99A3A4","#7F8C8D", "#707B7C", "#616A6B","#515A5A", "#424949", "#D6DBDF", "#AEB6BF", "#808B96","#EAEDED", "#D5DBDB", "#E5E7E9", "#D7DBDD","#CACFD2", "#BDC3C7", "#A6ACAF", "#909497", "#797D7F","#626567","#ABB2B9","#808B96","#EAECEE","#EBEDEF")
Treeplot function
Function to create a tree from a phyloseq object which contains a tree. With edges coloured by a certain taxa rank
# The taxarank has to be entered twice in this function: e.g. taxalevel = "Phylum", Phylum
treefunction <- function(phyloseqfile, layout = "circular", taxalevel = "Phylum", taxalevelforggplot) {
## Extract a taxadataframe based on your final filters
taxa.df <- phyloseq::tax_table(phyloseqfile)@.Data %>% as.data.frame
## Create a list to split otus by phyla, this will be used for colour-grouping the tree
taxa.df.phylum <- taxa.df %>% rownames_to_column("OTUID") %>% dplyr::select(OTUID, taxalevel) %>%
column_to_rownames("OTUID")
phyla.list <- split(rownames(taxa.df.phylum), as.vector(taxa.df %>% dplyr::select(taxalevel)), drop = TRUE)
## adding that phyla list to the tree
tree.df <- phyloseq::phy_tree(phyloseqfile)
### add list grouping to tree and call it Phylum (or another taxa level)
ggtree_gps <- ggtree::groupOTU(tree.df, phyla.list, taxalevel)
### Plot tree
ggtree::ggtree(ggtree_gps, aes(color={{taxalevelforggplot}}, alpha = 0.5), layout = layout) +
theme(legend.position="right") +
scale_colour_manual(values = treecols) +
guides(color = guide_legend(override.aes = list(size=5)))
}
Treeplot function phylofactor
This function produces a treeplot with phylofactors highlighted and includes a legend to identify them.
It is slightly modified from the original phylofactor function pf.tree.
# function for a tree figure with phylofactors highlighted including a legend to identify them
pf.tree.with.legend <- function(pf, layout = "circular", factorvector = NULL) {
factor.map=data.frame('Phylofactor'=1:pf$nfactors,'group'=rep(1,pf$nfactors))
if (is.null(factorvector)) {
factor.map=data.frame('Phylofactor'=1:pf$nfactors,'group'=rep(1,pf$nfactors))
} else {
factor.map <- factor.map %>% dplyr::filter(Phylofactor %in% factorvector)
}
m <- nrow(factor.map)
#color.fcn=viridis::viridis
#cols <- color.fcn(m)
#factor.map$colors <- cols
tree=pf$tree
n=ape::Ntip(tree)
method = "factors"
nd <- NULL
for (i in 1:m){
if (method=='factors'){
grp <- pf$tree$tip.label[pf$groups[[factor.map[i,1]]][[factor.map[i,2]]]]
} else {
grp <- pf$tree$tip.label[GroupList[[i]]]
}
grp <- intersect(grp,tree$tip.label)
if (length(grp)>0 & any(setdiff(tree$tip.label,grp) %in% pf$tree$tip.label)){
if (length(grp)>1){
nd <- c(nd, ggtree::MRCA(tree,grp))
} else {
nd <- c(nd,match(grp,tree$tip.label))
}
}
}
df <- data.frame(factor.map, node = nd)
df$Phylofactor <- factor(df$Phylofactor, levels = df$Phylofactor)
gg <- ggtree::ggtree(tree,layout=layout, aes(alpha = 0.5)) +
guides(alpha = FALSE)
gg <- gg+ggtree::geom_hilight(data = df, aes(node=node, fill=Phylofactor))
return(gg)
}
Add ILRs to phyloseq
Function to take a phylofactor file and extract the mean sample ILRs and add those to the phyloseq sample data They can then be used for plotting graphs together with abundance data.
addILRtophyloseq <- function(phyloseqobject, phylofactorobject) {
# export taxa data from phyloseq object
taxadf <- as.data.frame(tax_table(phyloseqobject)@.Data)
taxadf <- unite(taxadf, taxonomy, sep = ";", remove = TRUE, na.rm = FALSE) %>% rownames_to_column("OTU_ID")
# export metadata from phyloseq object
meta.df <- data.frame(sample_data(phyloseqobject)) %>% rownames_to_column("OTU_ID")
# add ILRs for each sample and factor to the metadataframe
factornumber <- nrow(phylofactorobject$factors)
for (i in 1:factornumber) {
smry <- phylofactor::pf.summary(phylofactorobject, taxadf, factor=i)
ilr.df <- data.frame(mean.ilr = smry$ilr) %>% rownames_to_column("OTU_ID")
meta.df <- meta.df %>% left_join(ilr.df, by = "OTU_ID")
names(meta.df)[ncol(meta.df)] <- paste0("meanILR_", i)
}
# re-import metadata into phyloseq object
phyloseqobject@sam_data <- phyloseq::sample_data(meta.df)
return(phyloseqobject)
}
Load phyloseq object
For detail on how to get a phyloseq object please see chapter ‘From Qiime2 into R’. It is important that the phyloseq object is the same phyloseq object that was used to extract the tree and ASV abundances to model the phylofactors.
# reading in a previously saved phyloseq object
physeqB.flt <- readRDS('physeqB.flt')
physeqB.flt
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 4932 taxa and 136 samples ]
## sample_data() Sample Data: [ 136 samples by 38 sample variables ]
## tax_table() Taxonomy Table: [ 4932 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 4932 tips and 4931 internal nodes ]
Filter phyloseq object as required for your anlaysis
It is important that the phyloseq object is the same phyloseq object that was used to extract the tree and ASV abundances to model the phylofactors.
`%notin%` = Negate(`%in%`)
# Filter out any sampled that are not needed for the analysis
physeqB.flt.norefs<- prune_samples(sample_data(physeqB.flt)$Treatment %notin% c("Reference", "Blank", "Heat"), physeqB.flt)
# Filter to specific sample categories
# Use a generic phyloseq object name - makes it less error-prone for downstream code
physeqB.flt.phyfac <- prune_samples(sample_data(physeqB.flt.norefs)$Week == "T52", physeqB.flt.norefs)
# remove any ASV that now have 0 abundance due to filtering
physeqB.flt.phyfac <- phyloseq::prune_taxa(taxa_sums(physeqB.flt.phyfac) != 0, physeqB.flt.phyfac)
physeqB.flt.phyfac
## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 3523 taxa and 40 samples ]
## sample_data() Sample Data: [ 40 samples by 38 sample variables ]
## tax_table() Taxonomy Table: [ 3523 taxa by 7 taxonomic ranks ]
## phy_tree() Phylogenetic Tree: [ 3523 tips and 3522 internal nodes ]
Load phylofactor object
See materials provided in resources to learn how to model phylofactors and create a phylofactor object.
# Read in the phylofactor object
PF_loss_T52_var_incH2O2 <- readRDS('PF_loss_T52_var_incH2O2')
PF_loss_T52_var_incH2O2
## phylofactor object from function PhyloFactor
## --------------------------------------------
## Method : glm
## Choice : var
## Formula : Data ~ X
## Number of species : 3523
## Number of factors : 58
## Frac Explained Variance : 0.00949
## Largest non-remainder bin : 2511
## Number of singletons : 33
## Paraphyletic Remainder : 818 species
##
## -------------------------------------------------------------
## Factor Table:
## Group1 Group2
## Factor 1 15 member Monophyletic clade 3508 member Monophyletic clade
## Factor 2 6 member Monophyletic clade 3502 member Paraphyletic clade
## Factor 3 2 member Monophyletic clade 3500 member Paraphyletic clade
## Factor 4 2511 member Paraphyletic clade 989 member Paraphyletic clade
## Factor 5 8 member Monophyletic clade 2503 member Paraphyletic clade
## Factor 6 5 member Monophyletic clade 2498 member Paraphyletic clade
## Factor 7 tip 2497 member Paraphyletic clade
## Factor 8 7 member Monophyletic clade 2490 member Paraphyletic clade
## Factor 9 tip 2489 member Paraphyletic clade
## Factor 10 10 member Monophyletic clade 979 member Paraphyletic clade
## ExpVar F Pr(>F)
## Factor 1 0.00063574 4.7144 0.03622200
## Factor 2 0.00049920 7.2506 0.01048600
## Factor 3 0.00035296 6.8365 0.01273500
## Factor 4 0.00036168 4.5507 0.03942300
## Factor 5 0.00036687 9.3068 0.00414820
## Factor 6 0.00035833 3.8421 0.05734500
## Factor 7 0.00033013 17.6360 0.00015554
## Factor 8 0.00029885 10.4280 0.00256100
## Factor 9 0.00028711 14.7260 0.00045600
## Factor 10 0.00025637 4.9842 0.03154900
Phylofactor analysis
Phylofactor summaries
Factor overview
The only difference to the factor overview from the generic output is that the coefficients of the model are shown. It helped me with interpretation of the effect-direction and strength.
# Factor overview
factors <- myfactoroverviewfunction(PF_loss_T52_var_incH2O2, physeqB.flt.phyfac)
library(kableExtra)
kable(head(factors), format = "html", digits = 2) %>% kable_styling(full_width = F, bootstrap_options = c( "condensed"), font_size = 10 )
| Factors | Group1 | Group2 | ExpVar | F | Pr..F. | (Intercept) | X |
|---|---|---|---|---|---|---|---|
| Factor 1 | 15 member Monophyletic clade | 3508 member Monophyletic clade | 0 | 4.71 | 0.04 | 4.24 | -0.32 |
| Factor 2 | 6 member Monophyletic clade | 3502 member Paraphyletic clade | 0 | 7.25 | 0.01 | -0.12 | 0.28 |
| Factor 3 | 2 member Monophyletic clade | 3500 member Paraphyletic clade | 0 | 6.84 | 0.01 | 0.75 | 0.24 |
| Factor 4 | 2511 member Paraphyletic clade | 989 member Paraphyletic clade | 0 | 4.55 | 0.04 | 5.33 | -0.24 |
| Factor 5 | 8 member Monophyletic clade | 2503 member Paraphyletic clade | 0 | 9.31 | 0.00 | 0.58 | 0.24 |
| Factor 6 | 5 member Monophyletic clade | 2498 member Paraphyletic clade | 0 | 3.84 | 0.06 | -0.95 | 0.24 |
Note: The model here found a total of 58 factors. Factor 4 contained the largest number of ASVs. What groups of bacteria were they? This can be explored numerically and visually.
Phylofactors by ASV
Each ASV is listed with phylofactor, significance, coeffients and abundances added as extra columns.
# Creating a phylofactor overview on ASV level
# Function as loaded earlier. Input the phylofactor object and the phyloseq object
## the third function input is '4' or '5', 4 is used when the phylofactor model setting 'choice' = 'F', otherwise use 5
phylfactor.taxa.summary <- phyfacsummary(PF_loss_T52_var_incH2O2, physeqB.flt.phyfac, 5)
kable(head(phylfactor.taxa.summary %>% dplyr::select(-OTU, -Kingdom)), format = "html", digits = 2) %>%
kable_styling(full_width = F, bootstrap_options = c( "condensed"), font_size = 10 )
| Phylum | Class | Order | Family | Genus | Species | Phylofactor | Pr(>F) | (Intercept) | X | Pr(>F)adj. | Abundance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chloroflexi | Ktedonobacteria | Ktedonobacterales | JG30-KF-AS9 | JG30-KF-AS9 | uncultured_soil | 1 | 0.04 | 4.24 | -0.32 | 0.04 | 310 |
| Chloroflexi | Ktedonobacteria | Ktedonobacterales | JG30-KF-AS9 | JG30-KF-AS9 | uncultured_soil | 1 | 0.04 | 4.24 | -0.32 | 0.04 | 300 |
| Chloroflexi | Ktedonobacteria | Ktedonobacterales | JG30-KF-AS9 | JG30-KF-AS9 | uncultured_soil | 1 | 0.04 | 4.24 | -0.32 | 0.04 | 317 |
| Chloroflexi | Ktedonobacteria | Ktedonobacterales | JG30-KF-AS9 | JG30-KF-AS9 | uncultured_soil | 1 | 0.04 | 4.24 | -0.32 | 0.04 | 606 |
| Chloroflexi | Ktedonobacteria | Ktedonobacterales | JG30-KF-AS9 | JG30-KF-AS9 | uncultured_soil | 1 | 0.04 | 4.24 | -0.32 | 0.04 | 14 |
| Chloroflexi | Ktedonobacteria | Ktedonobacterales | JG30-KF-AS9 | JG30-KF-AS9 | uncultured_soil | 1 | 0.04 | 4.24 | -0.32 | 0.04 | 456 |
Group by specified taxa level
Sometimes its easier to look at higher taxonomic levels first.
# create a summary of taxa by phylofactor and taxa level
# Group and sum abundances at order level
phylfactor.taxa.summary.taxalevel <- phylfactor.taxa.summary %>%
group_by(Phylum, Class, Order, Phylofactor) %>%
summarise(Abundance = sum(Abundance)) %>%
arrange(Phylofactor, Phylum)
kable(head(phylfactor.taxa.summary.taxalevel), format = "html", digits = 2) %>%
kable_styling(full_width = F, bootstrap_options = c( "condensed"), font_size = 10 )
| Phylum | Class | Order | Phylofactor | Abundance |
|---|---|---|---|---|
| Chloroflexi | Ktedonobacteria | Ktedonobacterales | 1 | 6890 |
| Proteobacteria | Gammaproteobacteria | Xanthomonadales | 2 | 8002 |
| Verrucomicrobiota | Verrucomicrobiae | Chthoniobacterales | 3 | 9160 |
| Acidobacteriota | Acidobacteriae | Acidobacteriales | 4 | 32861 |
| Acidobacteriota | Acidobacteriae | Bryobacterales | 4 | 3710 |
| Acidobacteriota | Acidobacteriae | Solibacterales | 4 | 1318 |
Filter specific factors if needed
# Summaries for speciec factors
phylfactor.taxa.summary.taxalevel <- phylfactor.taxa.summary %>%
group_by(Phylum, Order, Phylofactor) %>%
summarise(Abundance = sum(Abundance)) %>%
arrange(Phylofactor) %>%
filter(Phylofactor %in% c(4))
kable(head(phylfactor.taxa.summary.taxalevel), format = "html", digits = 2) %>%
kable_styling(full_width = F, bootstrap_options = c( "condensed"), font_size = 10 )
| Phylum | Order | Phylofactor | Abundance |
|---|---|---|---|
| Acidobacteriota | Acidobacteriales | 4 | 32861 |
| Acidobacteriota | Blastocatellales | 4 | 65 |
| Acidobacteriota | Bryobacterales | 4 | 3710 |
| Acidobacteriota | Pyrinomonadales | 4 | 8 |
| Acidobacteriota | Solibacterales | 4 | 1318 |
| Acidobacteriota | Subgroup_2 | 4 | 17440 |
Some other ways to look at phylofactors
# Sum of abundances of a specific phylofactor
sum((phylfactor.taxa.summary %>%
filter(Phylofactor %in% c(5)))$Abundance)
## [1] 8818
# AVS count of a specific phylofactor
nrow(phylfactor.taxa.summary %>%
filter(Phylofactor %in% c(5)))
## [1] 8
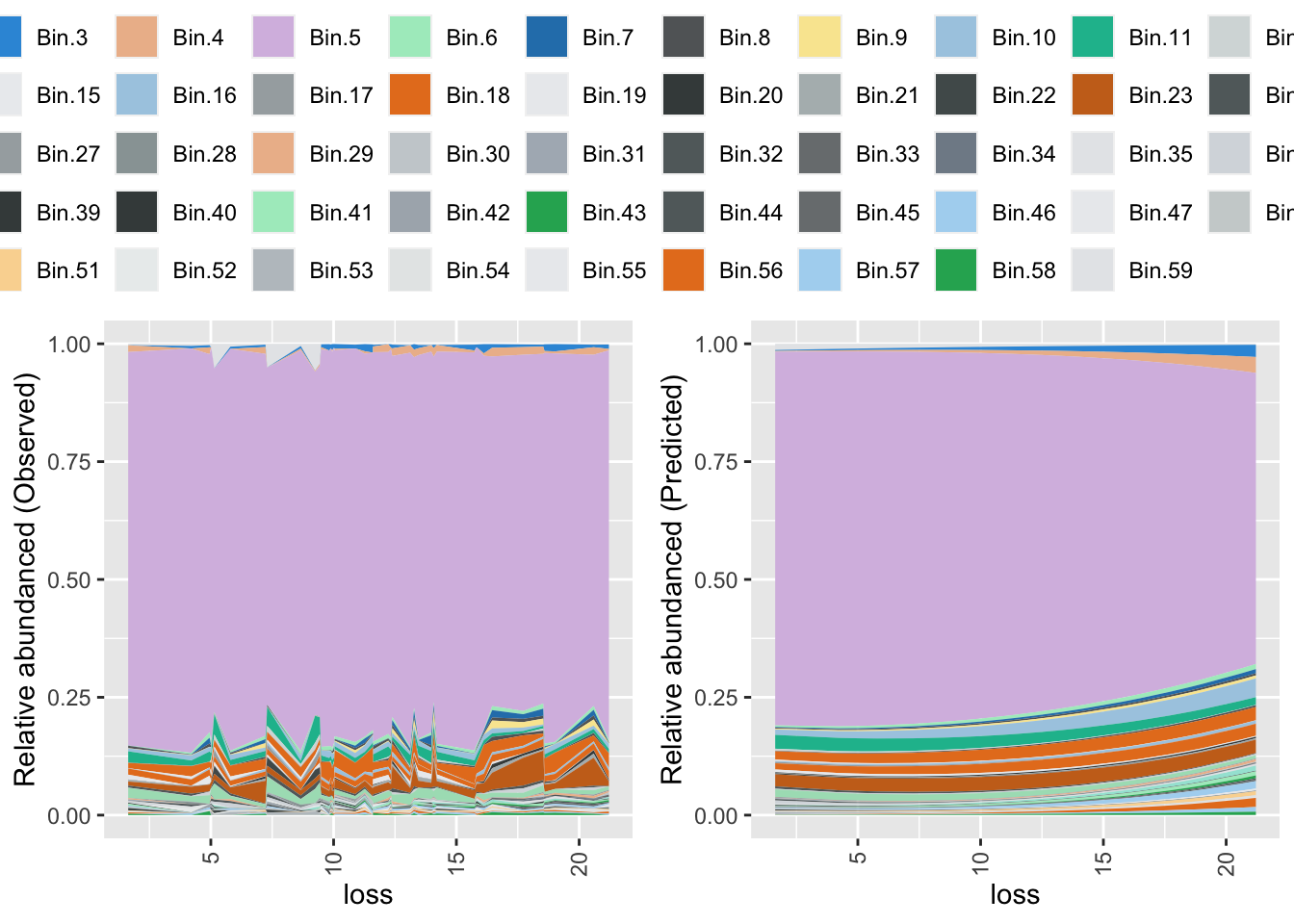
Plotting bins
This can help to get an idea of scale and direction of change effected by a continuous variable. However, I found analyzing bins mostly confusing.
# plotting bins
colvec <- mycolorfunction(PF_loss_T52_var_incH2O2, physeqB.flt.phyfac, loss, brightcols, greycols, 0.005, max)
binplotfunction(PF_loss_T52_var_incH2O2, physeqB.flt.phyfac, loss)
Plotting trees
Compare two differently coloured versions of the phylogenetic tree to assess patterns of change with your explanatory variable.
Tree 1 - colours indicate taxa that are associated with certain phylofactors
# Create colours (same as the pylofactors above) and name color vector by ILR name
# the name of the vector are required so ggpolot automatically allocated each colour to the right ILR
cols <- viridis::plasma(nrow(PF_loss_T52_var_incH2O2$factors))
# Name each colour in the colour vector (required for ggplot)
names(cols) <- data.frame('Phylofactor'=1:PF_loss_T52_var_incH2O2$nfactors,
'group'=rep(1,PF_loss_T52_var_incH2O2$nfactors))$Phylofactor
# Use the phylofactor tree function which we loaded earlier, highlighting a few pre-selected phylofactors
g.pf <- pf.tree.with.legend(PF_loss_T52_var_incH2O2, layout = "rectangular",
factorvector = c(2,3,4,5,8,17,27,28,29,39)) +
scale_fill_manual(values = cols)
#g.pf <- g.pf + ggtitle("Phylofactors")
g.pf
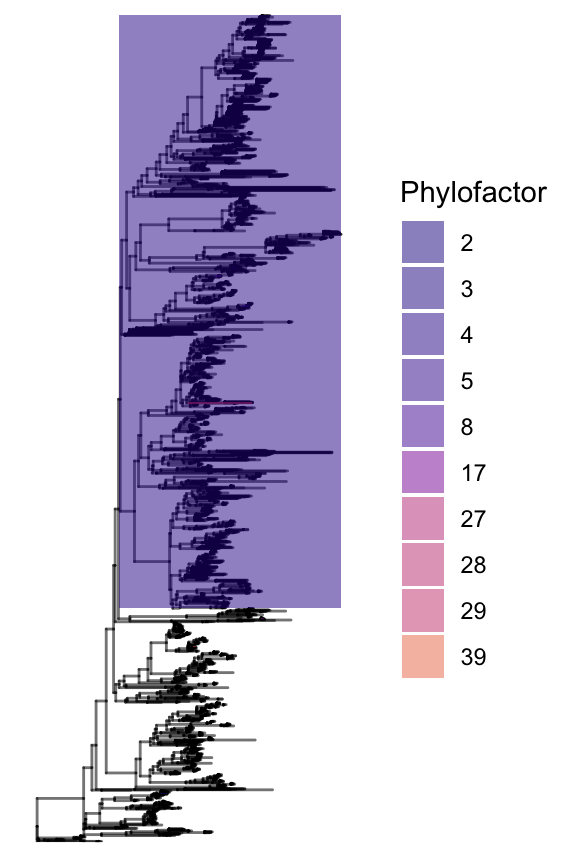
Note: Factor four consists of a large group of taxa (see the the purple box). This indicated that the variable ‘loss’ in this model was associated to abundances of a group of taxa that basically divided the phylogeny into two large groups. The next question was: What traits were associated to these two groups?
Also note that most of the other facors are difficult to see because they conistst of a smaller number of ASVs.
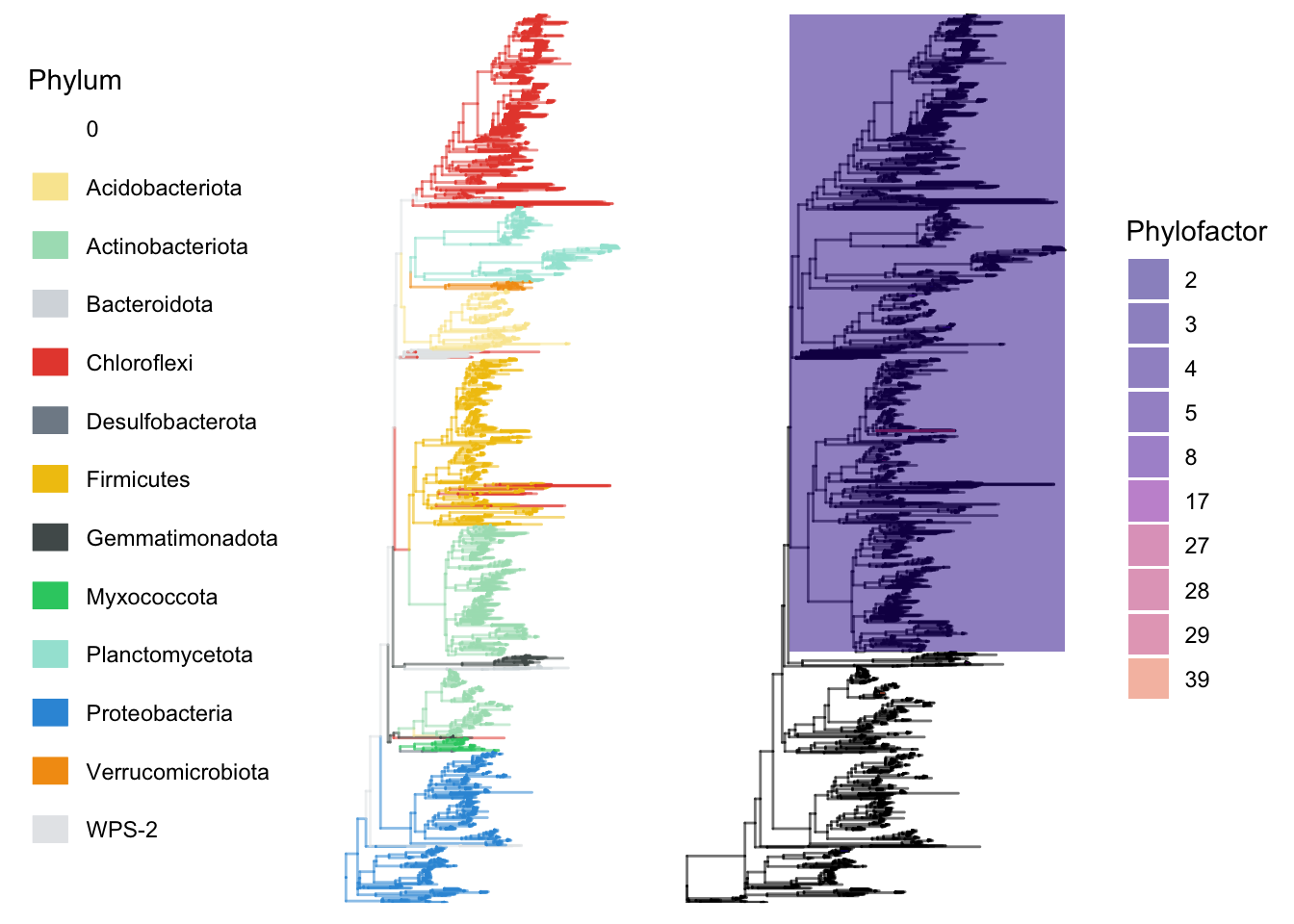
Tree 2 - coloured by phylum association
## Tree coloured by phylum
# Remove phyla that have low prevalence. These were identified in an earlier diagnostics in a previous chapter
physeqB.flt.phyfac.plotting <- physeqB.flt.phyfac %>%
subset_taxa(!Phylum %in%
c("RCP2-54", "Cyanobacteria","Dependentiae",
"Bdellovibrionota","Nitrospirota", "Armatimonadota",
"Armatimonadetes") )
physeqB.flt.phyfac.plotting = prune_taxa(taxa_sums(physeqB.flt.phyfac.plotting) != 0, physeqB.flt.phyfac.plotting)
# Use the tree functions loaded earlier
# get colour vector
treecols <- mytreecolorfunction(physeqB.flt.phyfac.plotting,
brightcols, greycols, 10000, 10, taxalevel =
"Phylum")
# The taxarank has to be entered twice in this function: e.g. taxalevel = "Phylum", Phylum
t <- treefunction(physeqB.flt.phyfac.plotting, layout = "rectangular",
taxalevel = "Phylum", Phylum) +
theme(legend.position = "left") +
guides(color = guide_legend(override.aes = list(size=5), ncol = 1, byrow = TRUE)) +
guides(alpha = FALSE)
#g2 <- ggtree::rotate_tree(gtree$ggplot,-45)
gg <- ggpubr::ggarrange(t, g.pf, widths = c(0.5,0.50))
gg
Add scatterplots of isometric log ratios (ILRs) vs. continous variable of interest.
# Use addILRtophyloseq function and create a new phyloseq object with added ILR results in the sample_data (for plotting)
physeqB.flt.ILR <- addILRtophyloseq(physeqB.flt.phyfac, PF_loss_T52_var_incH2O2)
# Perhaps do a quick Check that they have 'arrived'
# data.frame(sample_data(physeqB.flt.ILR))
# Extract plot data and pivot longer by ILR factors
plotdata <- data.frame(sample_data(physeqB.flt.ILR)) %>%
pivot_longer(cols = starts_with("meanILR"), names_to = "ILR")
# Make ILRs leveled factors so they appear in right order
plotdata$ILR <- factor(plotdata$ILR, levels =
plotdata[1:PF_loss_T52_var_incH2O2$nfactors,]$ILR)
# Create colours (same as the pylofactors above) and name color vector by ILR name
# the name of the vector are required so ggpolot automatically allocated each colour to the right ILR
cols <- viridis::plasma(nrow(PF_loss_T52_var_incH2O2$factors))
names(cols) <- plotdata[1:PF_loss_T52_var_incH2O2$nfactors,]$ILR
# plot ILRs
# I have pre-selected some factors for which I wanted to see the ILRs and compare
#or 1,2,3,4,5,6,8,9,10,14, 19,21, 23, 24,76,64,25,34,46,77,45
p1 <- plotdata %>% filter(ILR %in% c("meanILR_2","meanILR_3","meanILR_4","meanILR_5",
"meanILR_8","meanILR_17","meanILR_27","meanILR_28",
"meanILR_29","meanILR_39")) %>%
ggscatter(x = "loss", y = "value", combine = TRUE, add = "reg.line",
facet.by = "ILR", color = "ILR", ncol = 2,
add.params = list( color = "black") ,
ylab = c("Abundances (Isometric log ratio)"),
xlab = c("Dieldrin loss (%)")) +
scale_color_manual(values = c(cols)) +
theme(legend.position = "none") +
stat_cor(method = "spearman",
aes(label = paste(..rr.label.., ..p.label.., sep = "~`,`~")))
# combine with tree plots
gg1 <- ggarrange(gg, p1, widths = c(0.6,0.4))
#annotate_figure(gg1, top = text_grob("Sampled at T52 (excl. heat treatment), n = 40"))
## Save visualisation in publishable quality
#ggsave("TreePhylavsPF_plusILRs_T52.png", height=7, width=11, units='in', dpi=600)
knitr::include_graphics("./TreePhylavsPF_plusILRs_T52.png")
